Research
Our research is focused on several aspects of theoretical and computational phylogenetics. Here we list the main directions, but see the publications page for more details.
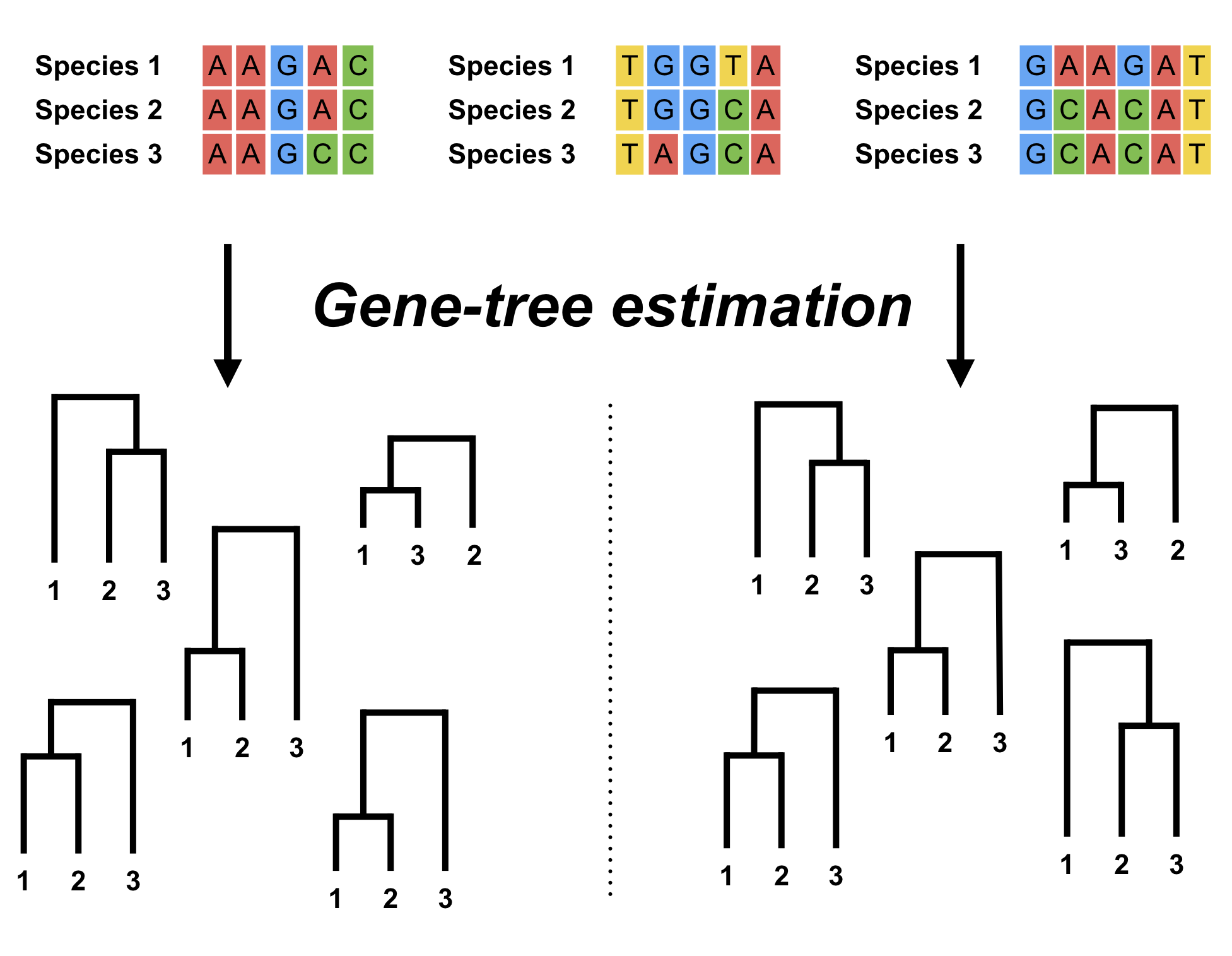
Robust Estimation of Gene Trees
Gene trees provide fundamental information about the processes in genetic evolution which is contained in the variation of their topology and branching times. In this work we are focused on how to reliably estimate gene trees by developing more robust statistical methods.
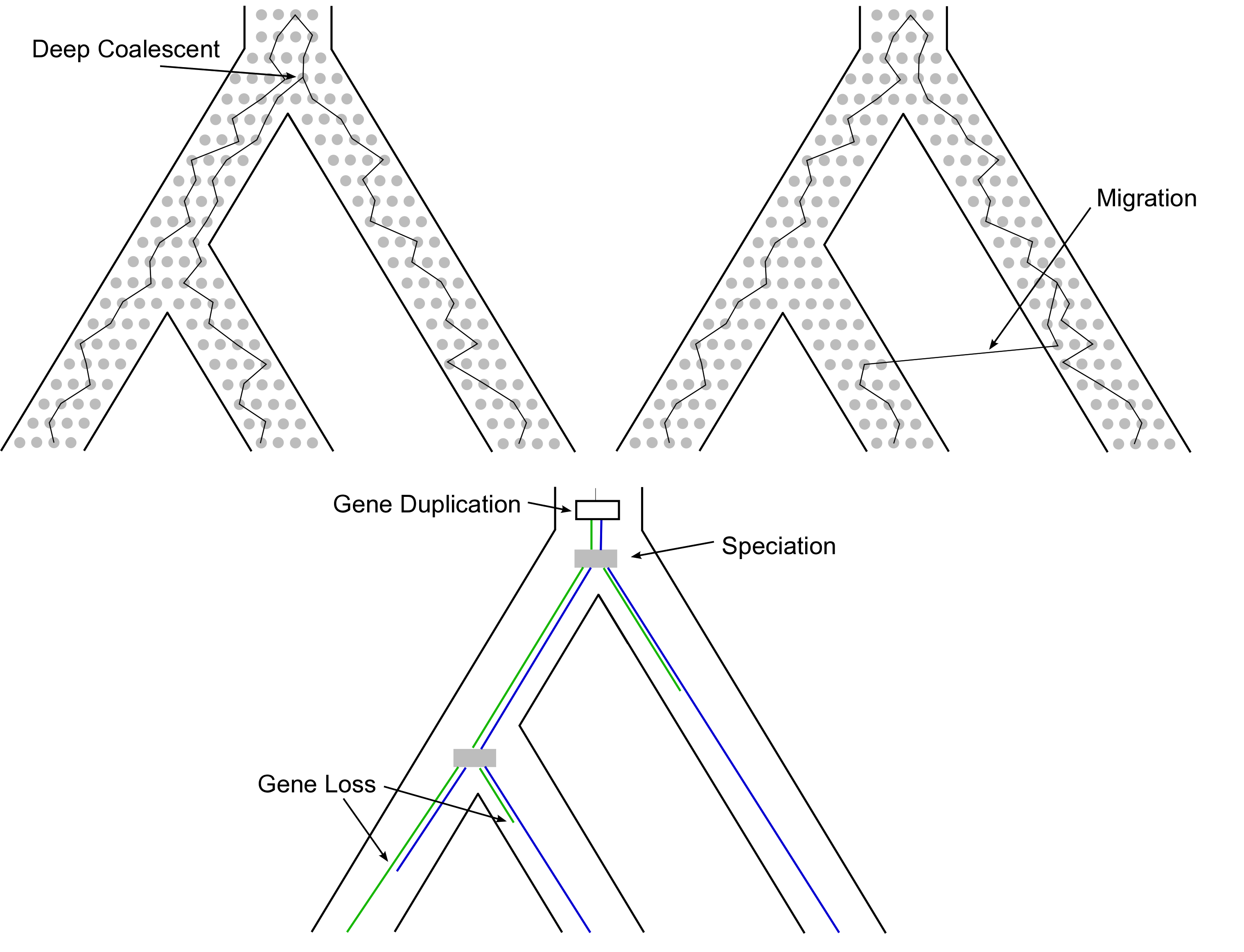
Understanding Discordance Between Species Trees and Gene Trees
The following biological scenarios can cause incongruent gene trees: (a) simple population genetic processes (i.e., the coalescent), (b) migration and thus gene-flow between species/populations , and (c) gene-duplication and gene-loss. We are working on more realistic models that incorporate different causes of gene-tree incongruence such as the multispecies coalescent with migration model. This work also entails new algorithm development and theory design for more efficient computation of species trees from thousands of gene trees.
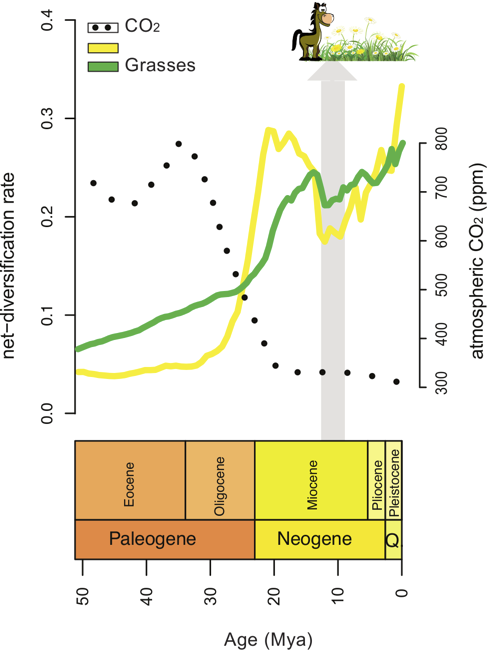
Inference of Macroevolutionary Processes
Our ultimate goal is to infer diversification rates through time and among lineages and identify correlations to genetic, phenotypic and/or environmental factors that impact diversification rate. For example, we developed a statistical method to identify correlations between rates of diversification and environmental variables, such as changes in atmospheric CO2. In our future work, we want to incorporate fossil occurrence information in our models of lineage diversification.
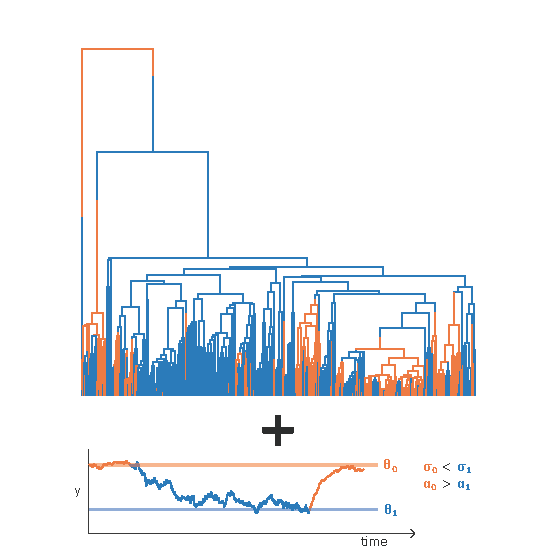
The State-Dependent OU Model
Joint inference of continuous trait adaptation and selective regime history using the state-dependent Ornstein-Uhlenbeck model. Trait evolution studies the evolution of homologous characters across phylogeny. These characters are at above-sequence level, including but not limited to gene expression, physiological, and morphological data. Many phylogenetic comparative methods (PCMs) have been developed to study trait evolution, yet to what extent these models reflect realistic evolutionary processes is still under debate. Here, we have developed the state-dependent Ornstein-Uhlenbeck model, a Bayesian approach to jointly infer the evolution of a continuous trait and the discrete selective regimes.
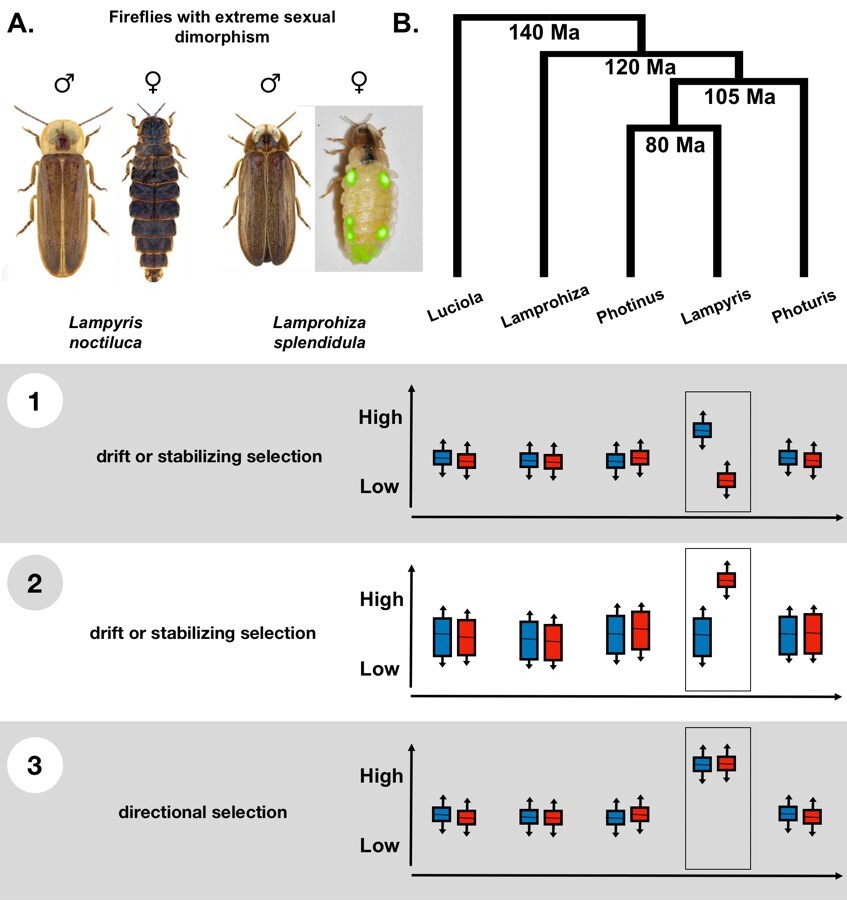
Modeling gene expression evolution in fireflies
Gene expression is a key driver of trait variation, particularly among closely related species. This project aims to develop innovative methods to model gene expression evolution using Brownian motion and Ornstein-Uhlenbeck processes. Specially, we focus on within-species variance, a critical yet underexplored aspect of gene expression. The firefly family (Lampyridae) serves as a novel study system due to their recurrent sexual dimorphism across the phylogeny. As sexual dimorphism is inherently linked to sex-biased gene expression, this makes fireflies an ideal model for investigating sex-biased gene expression evolution.

MacDrive
Biodiversity is modeled by the process of speciation and extinction. There is clear evidence from both living and extinct species that biodiversity is extremely variable through time and among species. However, we still do not know what factors drive speciation and extinction rates on a macroevolutionary level (that is, beyond species boundaries). The goal of this project is to combine statistical, computational, neontological – i.e. relating to species living today – and paleobiological approaches to study macroevolutionary dynamics.